

In silico comparative drug repurposing analysis of selected antifungal agents against SARS-CoV-2 main protease (Mpro, PDB ID: 6LU7): Molecular docking, ADMET profiling, and drug-likeness evaluation

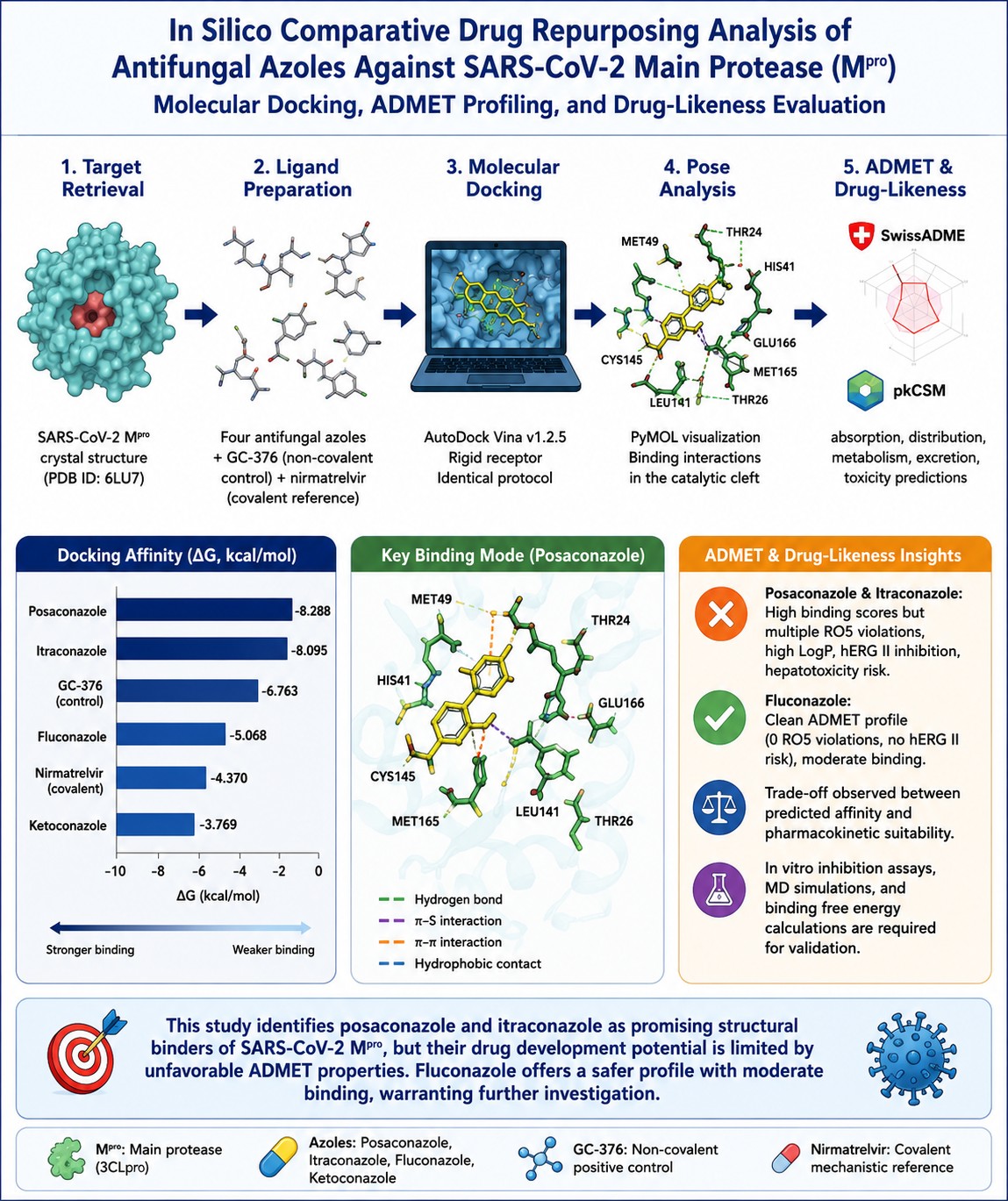
SARS-CoV-2 main protease (Mpro, 3CLpro, PDB ID: 6LU7) cleaves viral polyproteins pp1a and pp1ab at 11 conserved recognition sites, releasing the non-structural proteins needed for replication–transcription complex assembly. Its catalytic HIS41-CYS145 dyad lacks a close human homolog, providing Mpro with a reasonable selectivity window as a drug target. Four approved antifungal azoles—posaconazole, itraconazole, fluconazole, and ketoconazole—were evaluated in this study as candidate non-covalent inhibitors against the 6LU7 crystal structure (2.16 Å) using AutoDock Vina v1.2.5 under rigid-receptor conditions. GC-376, a non-covalent Mpro inhibitor with published IC50 data, was docked under identical conditions and served as the primary scoring reference (ΔG = −6.763 kcal/mol). Nirmatrelvir was retained solely for the mechanistic context; rigid non-covalent docking cannot model its CYS145-alkylating mechanism. Posaconazole returned the strongest predicted affinity (−8.288 kcal/mol), followed by itraconazole (−8.095 kcal/mol), fluconazole (−5.068 kcal/mol), nirmatrelvir (−4.370 kcal/mol), and ketoconazole (−3.769 kcal/mol). Posaconazole occupied the HIS41-CYS145 catalytic cleft with predicted contacts at MET49 (~3.8 Å), MET165 (~4.1 Å), GLU166 (~3.3 Å), and the catalytic dyad residues HIS41 (~3.2 Å) and CYS145 (~3.5 Å). These distances were estimated from a rigid-receptor protocol and carry inherent coordinate uncertainty; they are not experimentally validated geometries. Posaconazole and itraconazole both carry multiple Lipinski violations, elevated lipophilicity (LogP > 4.5), predicted hERG II inhibition, and hepatotoxicity risk. Fluconazole showed a clean absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile, with weaker predicted binding (−5.068 kcal/mol). No antiviral activity was claimed. Molecular dynamics simulation, binding free energy calculation, and in vitro Mpro inhibition assays are required before any biological inference can be drawn.
- Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270-273. doi: 10.1038/s41586-020-2012-7
- Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol. 2000;81(Pt 4):853-879. doi: 10.1099/0022-1317-81-4-853
- Hegyi A, Ziebuhr J. Conservation of substrate specificities among coronavirus main proteases. J Gen Virol. 2002;83(Pt 3):595-599. doi: 10.1099/0022-1317-83-3-595
- Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368(6489):409- 412. doi: 10.1126/science.abb3405
- Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289- 293. doi: 10.1038/s41586-020-2223-y
- Owen DR, Allerton CMN, Anderson AS, et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374(6575):1586- 1593. doi: 10.1126/science.abl4784
- Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18(1):41-59. doi: 10.1038/nrd.2018.168
- Hof H. A new, broad-spectrum azole antifungal: posaconazole—mechanisms of action and resistance, spectrum of activity. Mycoses. 2006;49(Suppl 1):2-6. doi: 10.1111/j.1439-0507.2006.01295.x
- Bruggemann RJM, Alffenaar JWC, Blijlevens NMA, et al. Clinical relevance of pharmacokinetic interactions of azole antifungal drugs with other coadministered agents. Clin Infect Dis. 2009;48(10):1441-1458. doi: 10.1086/598327
- Ghosh R, Chakraborty A, Biswas A, Chowdhuri S. Identification of polyphenols from Broussonetia papyrifera as SARS-CoV-2 main protease inhibitors using in silico docking and molecular dynamics simulation approaches. J Biomol Struct Dyn. 2021;39(17):6747-6760. doi: 10.1080/07391102.2020.1802347
- Ma C, Sacco MD, Hurst B, et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678-692. doi: 10.1038/s41422-020-0356-z
- Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6(11):881-890. doi: 10.1038/nrd2445
- Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235-242. doi: 10.1093/nar/28.1.235
- Liu X, Zhang B, Jin Z, Yang H, Rao Z. The crystal structure of COVID-19 main protease in complex with an inhibitor N3. RCSB PDB. 2020. doi: 10.2210/pdb6LU7/pdb
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785-2791. doi: 10.1002/jcc.21256
- Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49(D1):D1388-D1395. doi: 10.1093/nar/gkaa971
- O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33
- Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J Chem Inf Model. 2021;61(8):3891-3898. doi: 10.1021/acs.jcim.1c00203
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 2.5. New York, NY; 2021. https://www.pymol.org
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3- 26. doi: 10.1016/S0169-409X(00)00129-0
- Daina A, Zoete V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem Med Chem. 2016;11(11):1117-1121. doi: 10.1002/cmdc.201600182
- Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066- 4072. doi: 10.1021/acs.jmedchem.5b00104
- European Medicines Agency. Ketoconazole-containing medicines: referral. 2013. Accessed May 2026. https:// www.ema.europa.eu/en/medicines/human/referrals/ ketoconazole-containing-medicines
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440(7083):463-469. doi: 10.1038/nature04710
- Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449-461. doi: 10.1517/17460441.2015.1032936
- Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384-13421. doi: 10.3390/molecules200713384